
جلسه دفاع از رساله دکتری (آقای رضا شهسواری)
موضوع: شبیه سازی دینامیک مولکولی برای مطالعه رفتار مکانیکی تک لایه دیسولفید مولیبدن با استفاده از پتانسیل های بین اتمی یادگیری ماشین
استادان راهنما: دکتر سیدعلیرضا شهیدی، دکتر سید جواد هاشمی فر
استادان مشاور:
استادان داور: دکتر محمد سیلانی، دکتر مجتبی اعلایی
چیکده
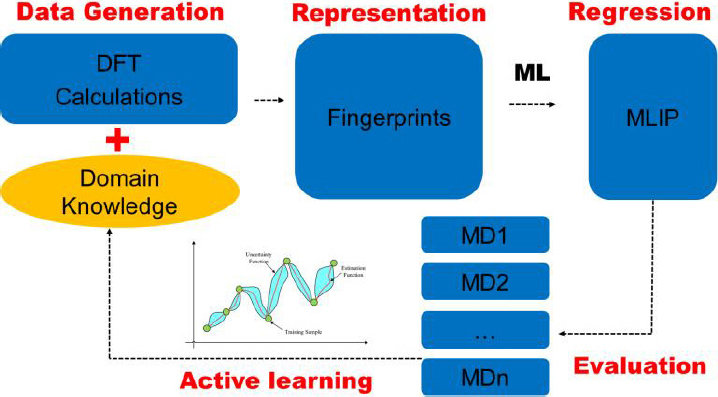
یکی از اهداف نهایی شبیه سازی های عددی، محاسبه دقیق خواص مواد با حداقل اطلاعات تجربی یا آزمایشگاهی می باشد. روش های ابتدا به ساکن مانند تئوری تابعی چگالی بهترین دقت ممکن را برای خواص الکترونیکی بدون نیاز به هیچ داده تجربی فراهم می کنند اما این روش برای سیستم هایی با صد یا نهایت هزار اتم کاربرد دارد. در واقع اگرچه محاسبات تئوری تابعی چگالی ابزار قدرتمندی برای تحقیق در مورد خواص مکانیکی و الکتریکی ساختارها می باشد اما برای سیستم های بزرگ از نظر محاسباتی به شدت هزینه بر می شوند. درحالی که اگرچه روش هایی مانند دینامیک مولکولی و یا اجزاء محدود برای سیستم های بزرگتر و واقعی تر کاربرد دارند ولی نیازمند داده های تجربی و آزمایشی هستند.
در این رساله، یک پتانسیل بین اتمی برای دیسولفید مولیبدن براساس تانسور پتانسیل ممان، که یکی از انواع پتانسیل های بین اتمی یادگیری ماشین می باشد ارائه می شود. در بخش به دست آوردن تابع پتاسیل یادگیری ماشین، نیروها، تنش ها و انرژی های تعدادی از ساختارهای دیسولفید مولیبدن، با استفاده از محاسبات تئوری تابع چگالی محاسبه خواهند شد تا یکسری داده های آموزشی فراهم شود. با توجه به دادههای آموزشی فراهم شده پتانسیل مورد نظر، با استفاده از پکیج پتانسیل های بین اتمی یادگیری ماشین که کارکرد آن براساس روش رگرسیون و الگوریتم غیرخطی می باشد آموزش داده خواهد شد تا بتواند پیش بینی را برای نیروها، تنش ها و انرژی های هر ساختار دیگری انجام دهد. بعد از به دست آوردن تابع پتانسیل بین اتمی یادگیری ماشین، یک شبیه سازی تست کشش تک محوره با استفاده از نرم افزار لمپس بر روی دیسولفید مولیبدن در هر دوفاز رسانا و نیمه رسانا انجام خواهد شد تا خواص مکانیکی این ماده شامل مدول یانگ، تنش حد نهایی، و کرنش شکست ارزیابی شوند.
کلمات کلیدی: دینامیک مولکولی، مکانیک کوانتومی، پتانسیل بین اتمی یادگیری ماشین، خواص مکانیکی، دیسولفید مولیبدن
